
 (Copia).jpg)

L’approvazione si basa su due studi positivi di Fase 3, EXPLORER-HCM e VALOR-HCM, che dimostrano un beneficio significativo nei pazienti trattati con mavacamten rispetto a placebo.
La Commissione Europea (EC) ha approvato mavacamten in capsule da 2,5 mg, 5 mg, 10 mg, 15 mg per il trattamento della cardiomiopatia ipertrofica (HCM) ostruttiva sintomatica (classe II-III NYHA – New York Heart Association) nei pazienti adulti. Mavacamten è il primo e unico inibitore selettivo allosterico e reversibile della miosina cardiaca approvato negli Stati membri* dell’Unione Europea (EU) e il primo inibitore di miosina cardiaca che interviene sui meccanismi fisiopatologici alla base della cardiomiopatia ipertrofica ostruttiva. L’approvazione della Commissione Europea di mavacamten si basa sui risultati positivi di efficacia e sicurezza degli studi di Fase 3 EXPLORER-HCM e VALOR-HCM.
La cardiomiopatia ipertrofica ostruttiva sintomatica è una malattia cardiaca spesso ereditaria, debilitante e progressiva e che può causare ai pazienti sintomi come mancanza di respiro, perdita di coscienza, vertigini e affaticamento, oltre a gravi complicazioni che alterano la vita, tra cui insufficienza cardiaca, aritmie, ictus e in rari casi (~ 1%), morte cardiaca improvvisa.
“La cardiomiopatia ipertrofica ostruttiva, spesso geneticamente determinata, causa un aumento dello spessore delle pareti del ventricolo sinistro del cuore – spiega Gianfranco Sinagra, Direttore Dipartimento Cardiotoracovascolare Azienda Sanitaria Universitaria Integrata di Trieste, Professore Ordinario di Cardiologia e Direttore della Scuola di Specializzazione in Malattie Apparato Cardiovascolare all’Università degli Studi di Trieste -. È caratterizzata da più componenti: l’ostruzione, cioè l’ostacolo all’uscita del sangue dal ventricolo sinistro, l’anomalia del rilasciamento, cioè la disfunzione diastolica e lo scompenso che ne può derivare, e l’alterata funzione della valvola mitrale. Vi è poi un secondo aspetto rappresentato dalle alterazioni dell’attività elettrica del cuore che deriva dalle cicatrici fibrotiche del miocardio, dall’alterata ossigenazione (ischemia) e da aritmie di vario tipo come la fibrillazione atriale. Nelle forme più gravi esiste il rischio di arresto cardiaco. Va ricordato che, pur essendo una patologia rara, la cardiomiopatia ipertrofica ostruttiva rientra tra le prime tre cause di morte improvvisa giovanile tra gli atleti. Le aritmie ventricolari pericolose per la vita possono verificarsi in persone giovani e negli atleti, senza essere precedute da sintomi premonitori. La malattia, infatti, può essere del tutto asintomatica, oppure presentare sintomi transitori come dispnea, affaticamento, perdita di coscienza. Gli esami previsti, soprattutto l’elettrocardiogramma, permettono di identificarla e di distinguerla da altre condizioni come il cosiddetto ‘cuore d’atleta’”. “È la malattia genetica familiare cardiaca più frequente, infatti in circa il 50-60% dei casi è geneticamente determinata – continua il Prof. Sinagra -. Questo permette di spostare l’attenzione dal paziente ai membri della famiglia consentendo diagnosi precoci ed approcci preventivi. L’identificazione della patologia ed il riscontro di mutazioni genetiche in un paziente determina l’avvio di programmi di screening anche nei familiari, con vari accertamenti come l’elettrocardiogramma, l’ecocardiogramma, in casi selezionati la risonanza magnetica cardiaca ed il test genetico. All’interno della famiglia, vi possono essere individui apparentemente sani, i cosiddetti ‘genotipo positivi’, cioè portatori della mutazione, ma con ‘fenotipo negativo’, cioè non affetti da evidenti variazioni dell’elettrocardiogramma ed ecocardiogramma. Queste persone devono essere sottoposte a controlli più ravvicinati, tesi ad evidenziare se nel tempo si manifesti lo sviluppo della malattia. La chirurgia è consigliata solo per le forme ostruttive gravi, non controllate dalla terapia farmacologica, e implica il ricovero in centri di eccellenza, perché è un intervento relativamente infrequente che richiede specifica esperienza e perizia. La maggior parte dei casi richiede una terapia farmacologica, che fino a oggi è stata costituita da betabloccanti, calcioantagonisti e disopiramide. L’approvazione di terapie come mavacantem è indubbiamente un importante passo avanti che consentirà di dare risposte concrete ai bisogni delle persone”.
“Si stima siano oltre 100mila le persone colpite da cardiomiopatia ipertrofica in Italia. Di queste, però, solo circa 15mila hanno probabilmente ricevuto una diagnosi corretta – afferma Iacopo Olivotto, Professore Ordinario di Cardiologia all’Università degli Studi di Firenze e Direttore della Cardiologia Pediatrica dell’Azienda Ospedaliero Universitaria Meyer IRCCS -. Il nostro Paese vanta una cultura decennale nella cura delle cardiopatie genetiche e i centri di riferimento per questa patologia sono diffusi su tutto il territorio. Tuttavia, nei casi più gravi, la cardiomiopatia ipertrofica ostruttiva richiede un expertise in specifiche terapie chirurgiche o interventistiche che può essere garantito solo da team multidisciplinari, che possiamo ribattezzare ‘HCM Heart Team’, presenti in pochi centri ad alta specializzazione. In questi pazienti, i risultati positivi dei due studi clinici di Fase 3 EXPLORER-HCM e VALOR-HCM mostrano l’efficacia di mavacamten nel migliorare la qualità della vita, il compenso emodinamico, la capacità di esercizio e il controllo dei sintomi, risultando di fatto una potenziale alternativa alla chirurgia”.
“Lo studio EXPLORER-HCM è una pietra miliare, perché è il primo trial che ha portato all’approvazione di un farmaco espressamente sviluppato per la cura della cardiomiopatia ipertrofica, una patologia finora orfana sul piano farmacologico – continua il Prof. Olivotto, che è Principal Investigator dello studio EXPLORER-HCM -. In questo studio, che ha coinvolto 251 pazienti con cardiomiopatia ipertrofica ostruttiva sintomatica, il 65% ha evidenziato il miglioramento di almeno una classe funzionale, cioè dei parametri utilizzati per quantificare i sintomi, la condizione fisica, la funzione sociale e la qualità di vita. Lo studio VALOR-HCM, inoltre, ha evidenziato che l’aggiunta di mavacamten riduce significativamente il bisogno di procedure invasive: al termine dello studio, l’82% dei pazienti trattati – inizialmente destinati alla chirurgia – non aveva più tale indicazione. Accanto a questi importanti dati di efficacia, mavacamten ha mostrato un eccellente profilo di sicurezza e tollerabilità. Ci auguriamo che, dopo l’approvazione europea, anche i pazienti del nostro Paese possano avere a disposizione questa terapia innovativa quanto prima”.
“Questa approvazione è un’importante pietra miliare per i pazienti in Europa che ora avranno a disposizione l’opzione terapeutica mavacamten, inibitore first-in-class della miosina cardiaca che colpisce la fisiopatologia alla base della cardiomiopatia ipertrofica ostruttiva sintomatica”, afferma Samit Hirawat, M.D., chief medical officer, Bristol Myers Squibb. “Siamo orgogliosi di offrire questo trattamento innovativo ad un numero maggiore di pazienti nel mondo e di confermare il nostro impegno a livello globale a trasformare la vita dei pazienti grazie alla scienza”.
Bristol Myers Squibb ringrazia i pazienti e i ricercatori coinvolti nei due studi clinici.
*L’autorizzazione alla commercializzazione centralizzata non comprende l’approvazione in Gran Bretagna (Inghilterra, Scozia, Galles).
Il Riepilogo europeo delle caratteristiche del prodottomavacamten è disponibile sul sito di EMA all’indirizzo www.ema.europa.eu
Lo studio EXPLORER-HCM
EXPLORER-HCM (NCT03470545) è uno studio di Fase 3 in doppio cieco, randomizzato, controllato con placebo, a gruppi paralleli che ha arruolato complessivamente 251 pazienti adulti con cardiomiopatia ipertrofica ostruttiva sintomatica (classe NYHA II o III). Tutti i partecipanti presentavano una frazione di eiezione ventricolare sinistra misurabile (LVEF) ≥55% e un gradiente di picco del tratto di efflusso ventricolare sinistro (LVOT) (a riposo e/o provocato alla diagnosi) con almeno un picco ≥50 mmHg; inoltre, allo screening era richiesto un gradiente LVOT Valsalva ≥30 mmHg.
Il 92% dei pazienti risultava in terapia di base con un betabloccante o un calcio-antagonista. Al basale, circa il 73% dei pazienti randomizzati era di classe NYHA II e il 27% di classe NYHA III. La LVEF mediana era del 74%, e il gradiente LVOT Valsalva mediano era 73 mmHg. Il Kansas City Cardiomyopathy Questionaire-23 (KCCQ-23) Clinical Summary Score (CSS) mediano al basale era 71.
L’endpoint primario era rappresentato da una valutazione funzionale composita, misurata a 30 settimane, o della percentuale dei pazienti con un miglioramento del picco del consumo di ossigeno (pVO2) ≥1.5 mL/kg/min insieme a un miglioramento di almeno una classe NYHA o il miglioramento di pVO2 ≥3.0 mL/kg/min senza alcun peggioramento della classe NYHA. Gli endpoint secondari principali comprendono l’impatto sul gradiente LVOT post-esercizio, su pVO2, e sulla classe secondo il Class and Kansas City Cardiomyopathy Questionnaire (KCCQ)* e l’Hypertrophic Cardiomyopathy Symptom Questionnaire (HCMSQ)† alla settimana 30.
Lo studio ha soddisfatto tutti gli endpoint primari e secondari con significatività statistica:
-
Alla settimana 30, il 37% (n=45/123) dei pazienti trattati con mavacamten ha soddisfatto l’endpoint primario composito, definito come percentuale di pazienti che hanno raggiunto un miglioramento del picco del consumo di ossigeno (pVO2) ≥1.5 mL/kg/min oltre al miglioramento di almeno una classe NYHA oppure il miglioramento di pVO2 ≥3.0 mL/kg/min e nessun peggioramento della classe NYHA, rispetto al 17% (n=22/128) dei pazienti trattati con placebo. La differenza è stata del 19,4% (95% CI: 8,67-30,13; p=0,0005).
-
In aggiunta, alla settimana 30 i pazienti trattati con mavacamten hanno ottenuto notevoli miglioramenti rispetto al gruppo placebo in tutti gli endpoint secondari, quali:
-
Variazione dal basale del gradiente di picco LVOT post-esercizio [-47 mmHg vs -10 mmHg, differenza -35 (95% CI: -43, -28; p<0,0001)]
-
Variazione dal basale di pVO2 [1,4 mL/kg/min vs -0,05 mL/kg/min; differenza 1,4 (95% CI: 0,6-2; p<0 ,0006)]
-
Numero (%) dei pazienti con miglioramento della classe NYHA ≥ 1 [80 (65%) vs 40 (31%); differenza del 34% (95% CI; 22%, 45%; p<0,0001)]
-
Variazione dal basale del KCCQ-23 CSS [14 vs 4; differenza 9 (95% CI: 5, 13); p<0,0001]
-
Variazione dal basale del punteggio HCMSQ Shortness of Breath (SoB) [-2,8 vs -0,9; differenza -1,8 (95% CI: -2.4, – 1.2); p<0,0001]
-
*Il KCCQ-23 CSS deriva dal Total Symptoms Score (TSS) e dal punteggio Physical Limitations (PL) del KCCQ-23. Il punteggio va da 0 a 100 e il punteggio più alto rappresenta un migliore stato di salute.
†Il punteggioHCMSQ SoB misura la frequenza e la gravità della mancanza di respiro. Il punteggio va da 0 a 18 e il punteggio più basso rappresenta una minore mancanza di respiro.
Lo studio VALOR-HCM
VALOR-HCM (NCT04349072) è uno studio di Fase 3 randomizzato, in doppio cieco, controllato con placebo, multicentrico su pazienti con HCM ostruttiva sintomatica (classe NYHA II-IV) che soddisfano i criteri delle linee guida per la riduzione del setto (SRT; gradiente LVOT ≥ 50 mmHg e classe NYHA III-IV, o classe II con sincope da sforzo o presincope) e candidati o presi in considerazione (entro gli ultimi 12 mesi) per una procedura invasiva. Lo studio ha arruolato 112 pazienti (età media 60 anni; 51% uomini; 93% ≥ classe NYHA III) randomizzati 1:1 a ricevere mavacamten o placebo. Al basale il 95% dei pazienti era in terapia con betabloccanti o calcio-antagonisti, disopiramide o una combinazione di questi. L’endpoint composito primario è rappresentato dalla percentuale di pazienti che decidevano di procedere con la SRT prima o alla settimana 16 o da quelli che rimanevano eleggibili alla SRT secondo le linee guida (gradiente LVOT ≥50 mmHg e classe NYHA III-IV o classe II con sincope da sforzo o presincope) alla settimana 16. Gli endpoint secondari chiave includono la variazione rispetto al basale sul gradiente LVOT dopo esercizio, sulla classe NYHA e Kansas City Cardiomyopathy Questionnaire (KCCQ) e sui biomarcatori alla settimana 16.
Lo studio ha soddisfatto tutti gli endpoint primari e secondari con significatività statistica:
-
I risultati hanno mostrato che mavacamten ha ridotto significativamente l’endpoint composito primario relativo alla decisione del paziente di procedere con la SRT prima o alla settimana 16 o i pazienti che sono rimasti eleggibili alla SRT (gradiente LVOT ≥ 50 mmHg e classe NYHA III-IV, o classe II con sincope da sforzo o presincope) alla settimana 16, con l’82% dei pazienti non più eleggibili alla procedura chirurgica o che hanno deciso di non procedere con la SRT dopo 16 settimane di trattamento. Solo 10 (17,9%) pazienti trattati con mavacamten vs 43 (76,8%) nel gruppo placebo hanno deciso di procedere con la SRT prima o alla settimana 16 o sono risultati eleggibili alla SRT alla settimana 16; differenza tra i trattamenti (95% CI) del 58,9% (44,0% – 73,9%); P<0,0001.
-
I risultati hanno inoltre mostrato che mavacamten ha soddisfatto gli endpoint secondari (variazione dal basale alla settimana 16) rispetto al gruppo placebo:
-
Variazione dal basale del gradiente di picco LVOT post-esercizio [-39,1 mmHg vs -1,8 mmHg; differenza -37,2 mmHg (95% CI: -48,1 – 26,2), P<0,0001]
-
Percentuale con un miglioramento di almeno una classe NYHA [62,5% vs 21,4%; differenza 41,1% (95% CI: 24,5% – 57,7%), P<0,0001]
-
Variazione dal basale di KCCQ-23 CSS [10,4 vs 1,8; differenza 9,5 (95% CI: 4,9 – 14), P<0,0001]
-
Variazione dal basale di N-terminale del peptide natriuretico di tipo B (NT-proBNP) [0,35 vs 1,13; differenza 0,33 (95% CI: 0,27 – 0,42), P<0,0001]
-
Variazione dal basale della troponina cardiaca I [0,5 vs 1,03; differenza 0,53 (95% CI: 0,41 – 0,70), P<0,0001]
-
Dati di sicurezza aggregati degli studi EXPLORER-HCM e VALOR-HCM
Le reazioni avverse più comunemente riportate dai 179 pazienti trattati con mavacamten nei due studi di Fase 3 erano vertigini (17%), dispnea (12%), disfunzione sistolica (5%) e sincope (5%). In questi studi clinici il 5% (9/179) dei pazienti nel gruppo con mavacamten ha riportato la riduzione reversibile della LVEF < 50% (mediana 45%: intervallo: 35-49%) durante il trattamento. Nel 56% (5/9) di questi pazienti, sono state osservate riduzioni senza altre manifestazioni cliniche. In tutti i pazienti trattati con mavacamten la LVEF si è ristabilita con l’interruzione di mavacamten e hanno completato lo studio in trattamento. La dispnea è stata riportata nel 12,3% dei pazienti trattati con mavacamten rispetto all’8,7% di quelli trattati con placebo. Nello studio EXPLORER-HCM, la maggior parte (67%) degli eventi di dispnea è stata riportata dopo l’interruzione di mavacamten, con il tempo mediano di insorgenza di 2 settimane (intervallo: 0,1 – 4,9) dopo l’ultima dose.
Mavacamten
Mavacamten è il primo e unico inibitore di miosina cardiaca approvato negli Stati Uniti, indicato per il trattamento negli adulti della cardiomiopatia ipertrofica ostruttiva (HCM) sintomatica di classe II-III secondo la New York Heart Association (NYHA), che migliora le capacità funzionali e i sintomi. Ha ricevuto l’approvazione regolatoria anche in Australia, Canada, Brasile, Svizzera, Macau, Corea del Sud e Singapore. Mavacamten è un modulatore allosterico reversibile della miosina cardiaca. Modula il numero di teste di miosina che possono interagire con l’actina (generatori di energia), riducendo così la probabilità di formazione di ponti incrociati che producono forza (sistolica) e residua (diastolica). L’eccessiva formazione di ponti incrociati di miosina actina e la disregolazione dello stato di super-rilassamento (super-relaxed) sono segni distintivi del meccanismo dell’HCM. Mavacamten sposta la miosina in uno stato di risparmio energetico, che può essere reclutato e super-rilassato. Nei pazienti con HCM, l’inibizione della miosina con mavacamten riduce l’ostruzione LVOT dinamica e migliora le pressioni di riempimento cardiaco.
La cardiomiopatia ipertrofica ostruttiva
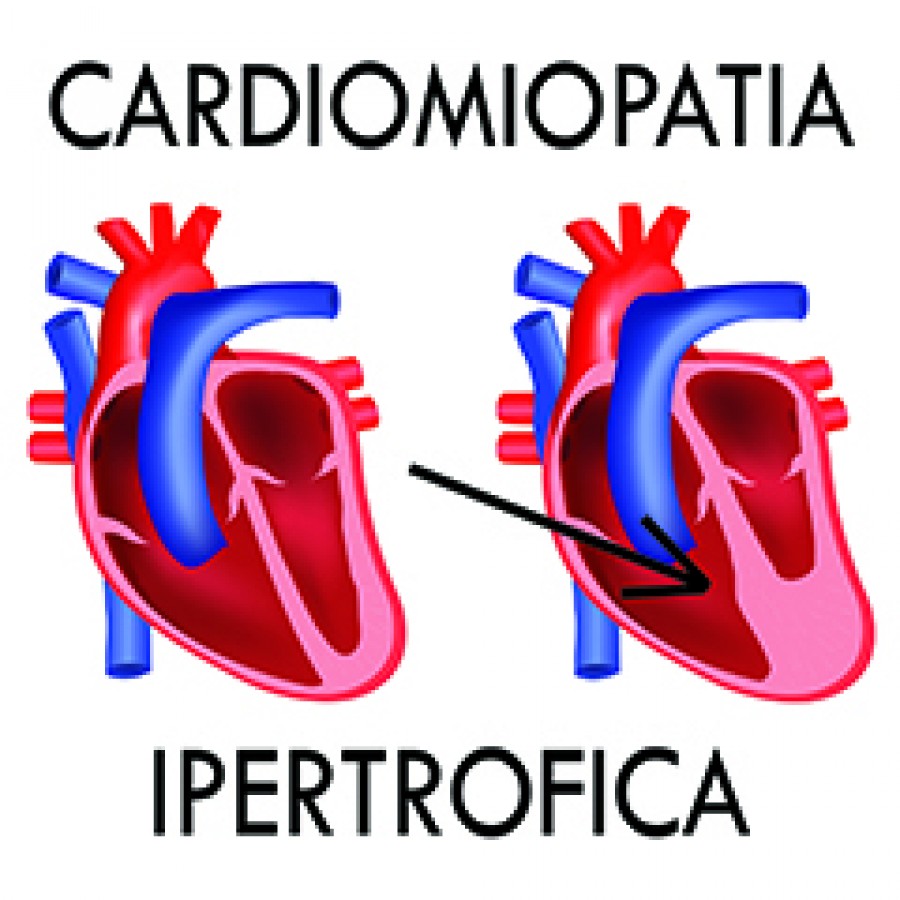
La cardiomiopatia ipertrofica (HCM) ostruttiva è una malattia cronica progressiva in cui l’eccessiva contrazione del muscolo cardiaco e la ridotta capacità di riempimento del ventricolo sinistro possono rendere difficile la circolazione del sangue nel corpo, causando lo sviluppo di sintomi debilitanti e disfunzione cardiaca. La cardiomiopatia ipertrofica può essere ereditaria e si può sviluppare a qualsiasi età. Viene diagnosticata generalmente a 40-50 anni e il 50% dei pazienti ha una predisposizione ereditaria.
Nella cardiomiopatia ipertrofica ostruttiva, il tipo più comune di HCM, l’efflusso ventricolare sinistro (LVOT) dove il sangue esce dal cuore risulta ostruito a causa dell’ingrossamento del muscolo cardiaco. Di conseguenza la cardiomiopatia ipertrofica ostruttiva è stata anche associata al rischio di fibrillazione atriale, infarto, insufficienza cardiaca e, anche se raramente, morte cardiaca improvvisa. La causa più frequente della cardiomiopatia ipertrofica è la mutazione delle proteine del muscolo cardiaco del sarcomero. Si stima che la cardiomiopatia ipertrofica ostruttiva colpisca 400.000-600.000 persone a livello mondiale, tuttavia in alcuni pazienti rimane non diagnosticata e/o asintomatica.
Bristol Myers Squibb
Bristol Myers Squibb è un’azienda biofarmaceutica globale la cui missione è scoprire, sviluppare e fornire farmaci innovativi che aiutino i pazienti a combattere malattie gravi. Per ulteriori informazioni su Bristol Myers Squibb BMS.com/it, LinkedIn, YouTube.

